 CHINESE / 中文语言
CHINESE / 中文语言  FRENCH / FRANCAIS
FRENCH / FRANCAIS  KOREAN / 한국어
KOREAN / 한국어  GERMAN / DEUTSCH
GERMAN / DEUTSCH  POLISH / JEZYK POLSKI
POLISH / JEZYK POLSKI  JAPANESE / NIHONGO
JAPANESE / NIHONGO  TURKISH / TÜRK DILI
TURKISH / TÜRK DILI  SPANISH / ESPAÑOL
SPANISH / ESPAÑOL  ITALIAN / ITALIANO
ITALIAN / ITALIANO 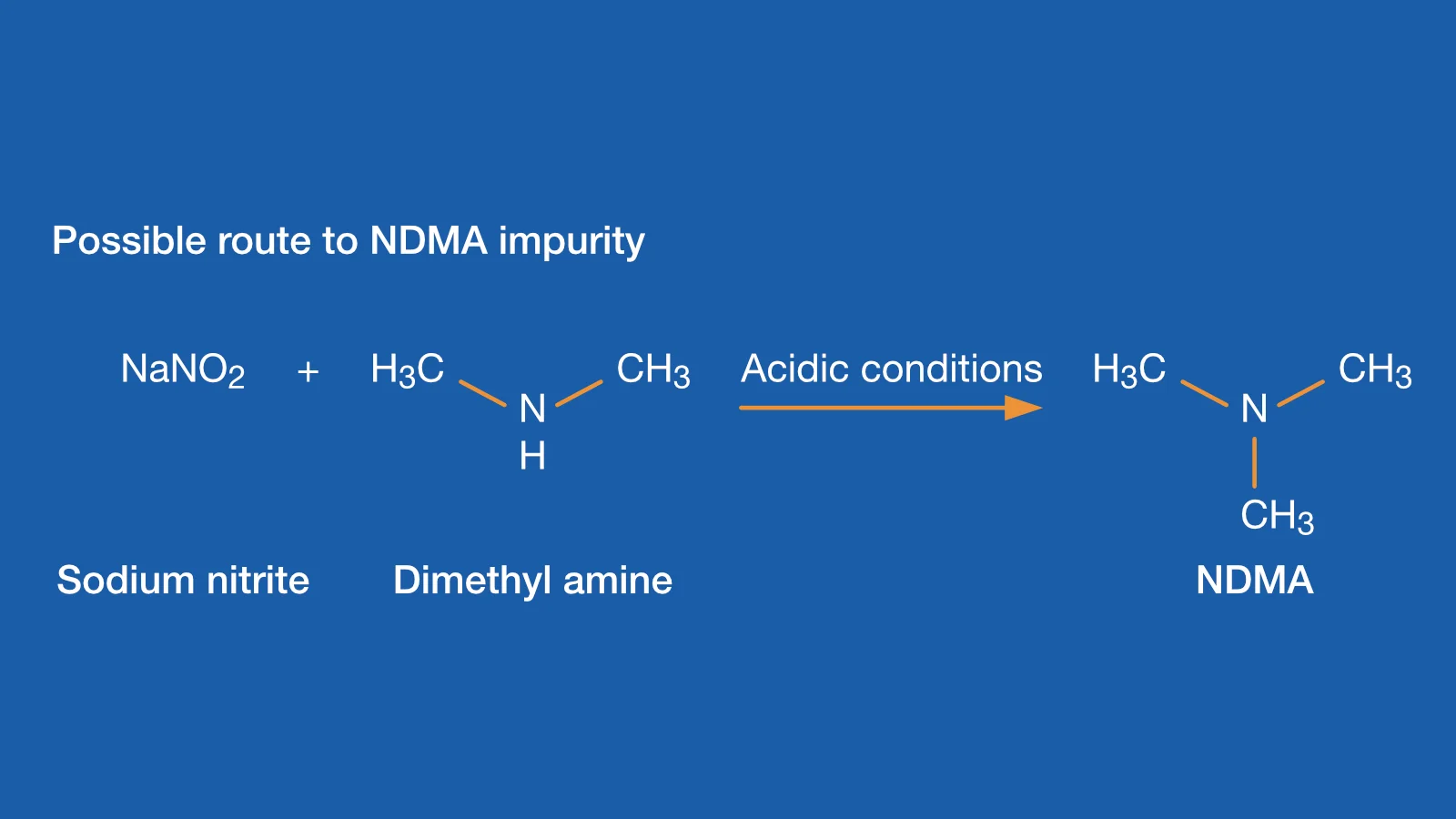
Identification and Quantification of Nitrosamines in Pharmaceutical Formulations.
Geneotoxic contaminants, such as Nitrosamines and their precursors, are a major concern for certain medicinal products. The US Food and Drug Administration (USFDA) and the European Medical Agency (EMA) require measurement of their concentration in chemically synthesized APIs.
There are many possible sources of these contaminants. These include raw materials, processing, packaging, environmental, and equipment to name just a few.
There is considerable debate over what the allowable limits of these compounds are and how the limit should be derived.
The EMS Assessment report EMA/369136/2020, 25 June 2020 states:
| The following limits have been established for some specific N-nitrosamines and should be applied: N-Nitrosamine (CAS number) | ng day*** |
|---|---|
| NDMA* (62-75-9) | 96.0 |
| NDEA*(55-18-5) | 26.5 |
| EIPNA**(16339-04-1) | 26.5 |
| DIPNA**(601-77-4) | 26.5 |
| NMBA**(61445-55-4) | 96.0 |
| MeNP**(16339-07-4) | 26.5 |
| NDBA**(924-16-3) | 26.5 |
These limits are applicable only if a finished product contains a single N-nitrosamine.
* Limit calculated on the basis of harmonic mean TD50 derived from carcinogenic potency database (CPDB)
**Limit derived using SAR/read-across approach
***The conversion to a specification limit in ppm for a particular medicinal product is calculated by dividing the respective above limit (ng) by the maximum daily dose (mg) of a given product as reflected in the SmPC.
The EMA assessment report focusses on LC-MS and GC-MS as being the correct methods for identifying and quantifying a wide range of nitrosamines. These methods are suitable for the detection of:
N-nitrosamines: N-nitroso-dimethylamine (NDMA, methods ABC); N-nitroso-diethylamine (NDEA, methods ABC); N-nitroso-dibutylamine (NDBA, method C); N-nitroso-N-methyl-4-aminoburyric acid (NMBA, method A); N-nitroso-diisopropylamine (NDiPA or DIPNA, methods AC); N-nitroso-ethyl-isopropylamine (NEiPA or EIPNA, methods AC) and N-nitroso-dipropylamine (NDPA, method C) in sartan-containing products and other formulations.
Method development and analysis services are available at MET to quantify these substances in your formulation. Studies can be applied to the final product in a validation scenario, batch release testing or as part of an investigation to identify sources of contamination.
Other contamination may also be introduced from degradation or from processing equipment. Analyses for these are equally available.
